This is the multi-page printable view of this section. Click here to print.
Blog
- Start Learning Bioinformatics
- Gene Download
- Amazing News For Gene Editing
- Test Dna Feature Viewer
- Edinburg Genome Foundry
- Human Hbb Gene
- Test Primer3 Py
Start Learning Bioinformatics
Learning bioinformatics was fun!
The start
I find a medical company near my home looking for bioinformatics engineer.
I first get some grasp on my Python web development, I even created a custom nginx package.
Then I’m stuck with caching. I use Firebase hosting instead.
After that I start using AI extensively, and crate a Web UI using deno was really easy.
But I want get some hands-on practice, so I discovered Syntheic Biology One.
The video
I learned so much! But when I recall I don’t remeber too much. weird. maybe I should watch again.
It’s produced near Germany. As always Germany gets you a rescue when you are in real trouble!
The end?
When I get some grasp I find that the job was closed. but I find there are also many companies looking for that position!
Then I discovered that there are still two lessons I don’t watched.
So maybe I will continue learning.
Side things
I watched Japanese porns by TakayaKi. As always Japanese warms your heart.
Also I watched Google IO in the meantime. The content was so full of use!
I also played King of Glory, but after so many days I find it finally boring.
Also PyCon US videos get released, so I guess I will start on that too.
But I don’t have too much money, so in the following month I guess I should look for a job while continue learning.
Gene Download
You’re absolutely on the right track with BioPython! It’s the go-to library for interacting with biological databases like NCBI programmatically in Python. Specifically, you’ll be using the Bio.Entrez module.
Here’s a breakdown of how to download a gene sequence from NCBI using BioPython, along with explanations and best practices:
Key BioPython Bio.Entrez Functions
-
Entrez.email: Crucial! You must set your email address before making any requests to NCBI. This helps NCBI understand who is using their E-utilities and allows them to contact you if there are issues. Without it, your requests might be blocked.
-
Entrez.api_key (Optional but Recommended): You can obtain an API key from your NCBI account. This allows you to make more requests per second (10 requests/second with a key vs. 3 requests/second without). For serious or frequent programmatic access, an API key is highly recommended to avoid hitting rate limits.
-
Entrez.esearch(): This function performs a search on an NCBI database (e.g., “nucleotide,” “gene,” “protein,” “pubmed”) and returns a list of matching IDs. Entrez.efetch(): Once you have the IDs, this function retrieves the full record(s) from the specified database in a desired format (e.g., FASTA, GenBank).
-
Entrez.read(): This function parses the XML output returned by esearch or efetch (when retmode=“xml”) into Python dictionaries and lists.
-
SeqIO.read() / SeqIO.parse(): From Bio.SeqIO, these functions are used to parse sequence files (or “handles” from efetch) into SeqRecord objects, which are very convenient for working with sequence data.
Step-by-Step Example: Downloading a Gene
Let’s say you want to download the sequence for the E. coli lacZ gene.
from Bio import Entrez
from Bio import SeqIO
import time # To respect NCBI's rate limits
# --- 1. Set your email and API key (if you have one) ---
# IMPORTANT: Replace with your actual email address
Entrez.email = "wjxa20152015@gmail.com"
# Optional: If you have an NCBI API key, uncomment and replace
# Entrez.api_key = "YOUR_NCBI_API_KEY"
# --- 2. Define your search query ---
# We'll search the 'gene' database first to get the gene ID,
# then use that to search the 'nucleotide' database for the sequence.
organism = "Escherichia coli"
gene_name = "lacZ"
search_term = f"{organism}[Organism] AND {gene_name}[Gene]"
print(f"Searching for gene: {search_term}")
# --- 3. Search for the gene ID in the 'gene' database ---
try:
handle = Entrez.esearch(db="gene", term=search_term, retmax="1") # retmax=1 to get the most relevant one
record = Entrez.read(handle)
handle.close()
gene_ids = record["IdList"]
if not gene_ids:
print(f"No gene ID found for '{search_term}'. Please check your search term.")
else:
ncbi_gene_id = gene_ids[0]
print(f"Found NCBI Gene ID: {ncbi_gene_id}")
# --- 4. Fetch the nucleotide sequence using the gene ID ---
# We need to specify the 'nucleotide' database for sequence data.
# rettype='fasta' for FASTA format, 'gb' for GenBank format.
# retmode='text' for plain text output.
print(f"Fetching nucleotide sequence for Gene ID: {ncbi_gene_id}")
# Pause to respect NCBI's rate limits (especially if not using API key)
time.sleep(0.5)
# For gene records, fetching the nucleotide sequence often involves searching
# the nucleotide database with the gene's locus tag or gene name in that context.
# A more robust way is to use the gene ID to find associated nucleotide records.
# Let's search the nucleotide database for the gene ID directly, or its accession.
# For simplicity, we'll try searching by gene name and organism again in 'nucleotide'
# or use the gene ID to link to a nucleotide entry if possible.
# Often, gene entries link to specific RefSeq accessions.
# A common approach: Use the Gene ID to find associated nucleotide records
# This can be tricky because a gene entry might link to a whole genome, not just the gene sequence.
# Let's try searching for the gene name and organism directly in the nucleotide database.
# This is often more straightforward for getting the actual gene sequence.
nucleotide_search_term = f"{organism}[Organism] AND {gene_name}[Gene] AND refseq[Filter]"
handle_nuc = Entrez.esearch(db="nucleotide", term=nucleotide_search_term, retmax="5")
nuc_record = Entrez.read(handle_nuc)
handle_nuc.close()
nucleotide_ids = nuc_record["IdList"]
if not nucleotide_ids:
print(f"No nucleotide sequence found for '{nucleotide_search_term}'. Trying broader search.")
# Fallback: if RefSeq filter didn't work, try without it
handle_nuc_broad = Entrez.esearch(db="nucleotide", term=f"{organism}[Organism] AND {gene_name}[Gene]", retmax="5")
nuc_record_broad = Entrez.read(handle_nuc_broad)
handle_nuc_broad.close()
nucleotide_ids = nuc_record_broad["IdList"]
if nucleotide_ids:
# Let's just take the first result for demonstration
target_nucleotide_id = nucleotide_ids[0]
print(f"Found Nucleotide ID: {target_nucleotide_id}")
# Fetch the sequence in FASTA format
time.sleep(0.5) # Respect rate limits
handle_fetch = Entrez.efetch(db="nucleotide", id=target_nucleotide_id, rettype="fasta", retmode="text")
fasta_sequence = handle_fetch.read()
handle_fetch.close()
print("\n--- FASTA Sequence ---")
print(fasta_sequence[:100] + '...')
# Optionally, parse it with SeqIO to get a SeqRecord object
from io import StringIO
seq_record = SeqIO.read(StringIO(fasta_sequence), "fasta")
print("\nSequence ID:", seq_record.id)
print("Sequence Length:", len(seq_record.seq))
print("First 50 bases:", seq_record.seq[:50])
# You can also save it to a file
output_filename = f"{seq_record.id}_{gene_name}.fasta"
with open(output_filename, "w") as out_handle:
out_handle.write(fasta_sequence)
print(f"\nSequence saved to {output_filename}")
# If you want GenBank format:
time.sleep(0.5) # Respect rate limits
handle_fetch_gb = Entrez.efetch(db="nucleotide", id=target_nucleotide_id, rettype="gb", retmode="text")
genbank_record = handle_fetch_gb.read()
handle_fetch_gb.close()
print("\n--- GenBank Record (first 500 chars) ---")
print(genbank_record[:500]) # Print first 500 characters
# You can parse the GenBank record with SeqIO as well:
# gb_seq_record = SeqIO.read(StringIO(genbank_record), "genbank")
# print("GenBank features:", gb_seq_record.features)
output_genbank_filename = f"{seq_record.id}_{gene_name}.gb"
with open(output_genbank_filename, "w") as out_handle_gb:
out_handle_gb.write(genbank_record)
print(f"GenBank record saved to {output_genbank_filename}")
else:
print("Could not find any nucleotide sequences for the specified gene.")
except Exception as e:
print(f"An error occurred: {e}")
Searching for gene: Escherichia coli[Organism] AND lacZ[Gene]
Found NCBI Gene ID: 945006
Fetching nucleotide sequence for Gene ID: 945006
Found Nucleotide ID: 2195797308
--- FASTA Sequence ---
>NZ_CAKNDS010000021.1 Escherichia coli strain cpe078, whole genome shotgun sequence
TGGCGGCTAATGGCAA...
Sequence ID: NZ_CAKNDS010000021.1
Sequence Length: 20027
First 50 bases: TGGCGGCTAATGGCAAGAAAGGAAAGGTGATTCTCGGCGCGATGATGCGC
Sequence saved to NZ_CAKNDS010000021.1_lacZ.fasta
--- GenBank Record (first 500 chars) ---
LOCUS NZ_CAKNDS010000021 20027 bp DNA linear CON 21-MAY-2025
DEFINITION Escherichia coli strain cpe078, whole genome shotgun sequence.
ACCESSION NZ_CAKNDS010000021 NZ_CAKNDS010000000
VERSION NZ_CAKNDS010000021.1
DBLINK BioProject: PRJNA224116
BioSample: SAMEA6451093
Assembly: GCF_929618365.1
KEYWORDS WGS; RefSeq.
SOURCE Escherichia coli
ORGANISM Escherichia coli
Bacteria; Pseudomonadati; Pseudomonadota; Gammaproteobact
GenBank record saved to NZ_CAKNDS010000021.1_lacZ.gb
from pathlib import Path
file = list(Path('.').glob("*.fasta"))[0]
content = file.read_text(encoding='utf8')
print(content[:100] + '...')
>NZ_CAKNDS010000021.1 Escherichia coli strain cpe078, whole genome shotgun sequence
TGGCGGCTAATGGCAA...
from Bio.Blast import NCBIWWW
from Bio.Blast import NCBIXML
from Bio import SeqIO
from io import StringIO
import time
# --- 1. Get your query sequence ---
# For demonstration, let's assume you have a FASTA string of your downloaded gene.
# In a real scenario, you would have loaded this from your downloaded .fasta file.
# Let's use the lacZ gene from the previous example for consistency.
# Replace this with your actual gene sequence (e.g., from reading the downloaded .fasta file)
lacZ_fasta_string = content
# For a real file:
# with open("your_gene.fasta", "r") as f:
# query_sequence_fasta = f.read()
# --- 2. Perform the BLAST search ---
print("Performing BLAST search... This may take a moment.")
# Example for nucleotide sequence (blastn)
# For protein sequences, you'd use 'blastp' or 'blastx' (nucleotide to protein)
# db: The database to search against (e.g., 'nr' for non-redundant, 'nt' for nucleotide, 'refseq_rna', 'refseq_genomic')
# You can find available databases on the NCBI BLAST website.
# Use a common database like 'nr' (nucleotide non-redundant) or 'nt' (nucleotide) for general searches.
# filter: 'L' for low complexity filter (recommended for most searches to avoid spurious hits)
try:
result_handle = NCBIWWW.qblast(
program="blastn", # or 'blastp', 'blastx', 'tblastn', 'tblastx'
database="nr", # or 'nt', 'refseq_rna', 'refseq_genomic', etc.
sequence=lacZ_fasta_string,
# Set a few more parameters if needed
# gapopen=11,
# gapextend=1,
# expect=10.0, # Expectation value (E-value) cutoff
# hitlist_size=10, # Number of descriptions and alignments to show
# format_type="XML" # The default format for NCBIXML parsing
)
# Pause to respect NCBI's rate limits (especially after a long query)
time.sleep(5) # A longer pause is often good after submitting a query
except Exception as e:
print(f"An error occurred during BLAST query: {e}")
print("Consider adding Entrez.email and Entrez.api_key if you haven't already.")
exit()
# --- 3. Parse the BLAST results ---
# The results are returned in XML format, which BioPython's NCBIXML can parse.
print("Parsing BLAST results...")
blast_records = NCBIXML.parse(result_handle) # parse() for multiple query sequences
# For a single query, you can use read():
# blast_record = NCBIXML.read(result_handle)
# Iterate through the results (there might be multiple records if you queried multiple sequences)
n = 0
for blast_record in blast_records:
print(f"\nQuery: {blast_record.query}")
print(f"Database: {blast_record.database}")
print(f"Number of hits found: {len(blast_record.alignments)}")
if not blast_record.alignments:
print("No significant alignments found.")
continue
# Iterate through the alignments (hits)
for alignment in blast_record.alignments:
for hsp in alignment.hsps: # HSP = High-scoring Segment Pair
if hsp.expect < 1e-10: # Filter by E-value (lower is better, closer to 0)
n +=1
if n >= 10: # first 10 results
break
print(f"\n Alignment: {alignment.title}")
print(f" Length: {alignment.length}")
print(f" E-value: {hsp.expect}")
print(f" Score: {hsp.score}")
print(f" Identities: {hsp.identities}/{hsp.align_length} ({hsp.positives} positives)")
print(f" Query start: {hsp.query_start}, end: {hsp.query_end}")
print(f" Subject start: {hsp.sbjct_start}, end: {hsp.sbjct_end}")
# You can print the actual alignment if needed
# print(f" Query: {hsp.query[0:75]}...")
# print(f" Match: {hsp.match[0:75]}...")
# print(f" Sbjct: {hsp.sbjct[0:75]}...")
break # Only process the first query for this example
result_handle.close()
print("\nBLAST search complete.")
Performing BLAST search... This may take a moment.
Parsing BLAST results...
Query: NZ_CAKNDS010000021.1 Escherichia coli strain cpe078, whole genome shotgun sequence
Database: core_nt
Number of hits found: 50
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 0.0
Score: 40054.0
Identities: 20027/20027 (20027 positives)
Query start: 1, end: 20027
Subject start: 3547756, end: 3567782
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 1.66533e-119
Score: 497.0
Identities: 280/298 (280 positives)
Query start: 1, end: 292
Subject start: 1600979, end: 1600682
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 7.08116e-118
Score: 490.0
Identities: 274/292 (274 positives)
Query start: 1, end: 292
Subject start: 3567840, end: 3567555
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 1.05094e-115
Score: 482.0
Identities: 241/241 (241 positives)
Query start: 1, end: 241
Subject start: 3919623, end: 3919863
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 3.21768e-103
Score: 437.0
Identities: 261/289 (261 positives)
Query start: 19758, end: 20027
Subject start: 1600633, end: 1600921
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 2.16997e-86
Score: 374.0
Identities: 216/234 (216 positives)
Query start: 19800, end: 20027
Subject start: 3548047, end: 3547814
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 1.3694e-82
Score: 360.0
Identities: 192/200 (192 positives)
Query start: 16847, end: 17046
Subject start: 3564402, end: 3564601
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 1.3694e-82
Score: 360.0
Identities: 192/200 (192 positives)
Query start: 16647, end: 16846
Subject start: 3564602, end: 3564801
Alignment: gi|1915194479|dbj|AP022409.1| Escherichia coli STW0522-31 DNA, complete genome
Length: 4783805
E-value: 4.77968e-82
Score: 358.0
Identities: 182/184 (182 positives)
Query start: 19844, end: 20027
Subject start: 3919864, end: 3919681
BLAST search complete.
from Bio import SeqIO
from dna_features_viewer import BiopythonTranslator
import matplotlib.pyplot as plt
# --- 1. Load the GenBank file into a Biopython SeqRecord object ---
# Replace 'your_gene.gb' with the actual path to your downloaded GenBank file
try:
record = SeqIO.read("NZ_CAKNDS010000021.1_lacZ.gb", "genbank")
print(f"Successfully loaded GenBank file: {record.id} ({len(record.seq)} bp)")
except FileNotFoundError:
print("Error: 'NZ_CAKNDS010000021.1_lacZ.gb' not found. Please make sure the file exists.")
# You might want to add code here to re-download the file if it's missing.
exit()
except Exception as e:
print(f"Error loading GenBank file: {e}")
exit()
# --- 2. Translate the Biopython record into a DNA Features Viewer GraphicRecord ---
# BiopythonTranslator automatically converts GenBank features into GraphicFeature objects.
graphic_record = BiopythonTranslator().translate_record(record)
# --- 3. Plot the graphic record ---
# This will generate a linear plot of your sequence with its features.
# 'figure_width' controls the width of the plot in inches.
ax, _ = graphic_record.plot(figure_width=10)
# --- 4. Add a title to your plot (optional) ---
ax.set_title(f"Features of {record.id}")
# --- 5. Display or save the plot ---
plt.tight_layout() # Adjusts plot to prevent labels from overlapping
plt.show() # Displays the plot
# To save the plot to a file instead of displaying:
# plt.savefig("gene_features_linear.png", dpi=300)
# plt.close() # Close the figure to free up memory
Successfully loaded GenBank file: NZ_CAKNDS010000021.1 (20027 bp)
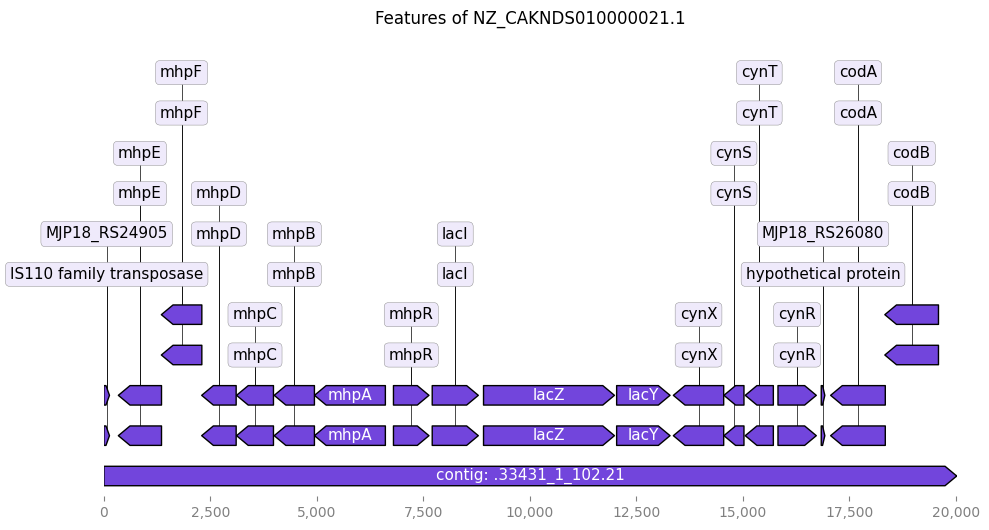
Test Dna Feature Viewer
from dna_features_viewer import GraphicFeature, GraphicRecord
features=[
GraphicFeature(start=0, end=20, strand=+1, color="#ffd700",
label="Small feature"),
GraphicFeature(start=20, end=500, strand=+1, color="#ffcccc",
label="Gene 1 with a very long name"),
GraphicFeature(start=400, end=700, strand=-1, color="#cffccc",
label="Gene 2"),
GraphicFeature(start=600, end=900, strand=+1, color="#ccccff",
label="Gene 3")
]
record = GraphicRecord(sequence_length=1000, features=features)
record.plot(figure_width=5)
(<Axes: >,
({GF(Small feature, 0-20 (1)): 0,
GF(Gene 1 with a very long name, 20-500 (1)): 0,
GF(Gene 2, 400-700 (-1)): 1.0,
GF(Gene 3, 600-900 (1)): 0},
{GF(Gene 1 with a very long name, 20-500 (1)): {'feature_y': 0,
'annotation_y': 2.0},
GF(Small feature, 0-20 (1)): {'feature_y': 0, 'annotation_y': 1.0}}))
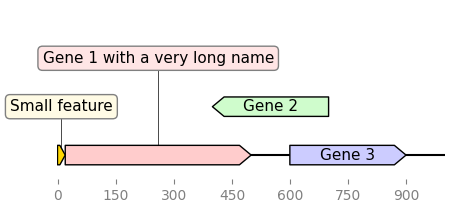
Edinburg Genome Foundry
https://edinburgh-genome-foundry.github.io/
This group build the tool: DNA Feature Viewer.
Some other tools are also intersting.
DNA Cauldron
DNA Cauldron (full documentation here) is a generic cloning simulation framework to predict final construct sequences and detect assembly flaws. It aims in particular at automating the simulation and verification of large (and possibly multi-step) DNA assembly batches, with the idea that, if your assembly plan works with Cauldron, you’ll get the same results at the bench.
Great support for Golden Gate assembly (incl. MoClo, EMMA, Phytobrick, etc.), the simulation is as simple as dropping sequence files in a web app.
Good support for Gibson, Biobrick, BASIC, LCR Assembly.
Provide genetic parts in any order, with or without annotations, in reverse or direct sense, linear or circular, single or combinatorial assembly… Cauldron will get it!
Design flaw detection (missing parts, unwanted restriction sites, wrong overhang designs, etc.).
If your assembly needs connector parts, Cauldron can select them for you!
Import batch assembly plans from spreadsheets, including hierarchical (=multi-step) plans.
Comprehensive reports with constructs sequences, fragment sequences, summary spreadsheets.
Export multi-file reports in folders, zip files, or in-memory zip files (for use on servers).
Web UI
They also built web UIs for their tools. for example:
Human Hbb Gene
https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=3043
Of course we can download it from the web page, but I’d like to download it via automated tools
BioPython can access Entrez database:
https://biopython.org/docs/latest/Tutorial/chapter_entrez.html
Test Primer3 Py
Project 2:
Primer Design Tool: Develop a tool (using Python and Primer3-py) that takes a DNA sequence as input, allows the user to specify target regions, and designs PCR primers.
Install
try:
from primer3 import bindings # type: ignore
except ImportError:
%pip install primer3-py
from primer3 import bindings # type: ignore
follow tutorial: primer design workflow
https://libnano.github.io/primer3-py/quickstart.html#workflow
r = bindings.design_primers(
seq_args={
'SEQUENCE_ID': 'MH1000',
'SEQUENCE_TEMPLATE': 'GCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCCTACATTTT'
'AGCATCAGTGAGTACAGCATGCTTACTGGAAGAGAGGGTCATGCA'
'ACAGATTAGGAGGTAAGTTTGCAAAGGCAGGCTAAGGAGGAGACG'
'CACTGAATGCCATGGTAAGAACTCTGGACATAAAAATATTGGAAG'
'TTGTTGAGCAAGTNAAAAAAATGTTTGGAAGTGTTACTTTAGCAA'
'TGGCAAGAATGATAGTATGGAATAGATTGGCAGAATGAAGGCAAA'
'ATGATTAGACATATTGCATTAAGGTAAAAAATGATAACTGAAGAA'
'TTATGTGCCACACTTATTAATAAGAAAGAATATGTGAACCTTGCA'
'GATGTTTCCCTCTAGTAG',
'SEQUENCE_INCLUDED_REGION': [36,342]
},
global_args={
'PRIMER_OPT_SIZE': 20,
'PRIMER_PICK_INTERNAL_OLIGO': 1,
'PRIMER_INTERNAL_MAX_SELF_END': 8,
'PRIMER_MIN_SIZE': 18,
'PRIMER_MAX_SIZE': 25,
'PRIMER_OPT_TM': 60.0,
'PRIMER_MIN_TM': 57.0,
'PRIMER_MAX_TM': 63.0,
'PRIMER_MIN_GC': 20.0,
'PRIMER_MAX_GC': 80.0,
'PRIMER_MAX_POLY_X': 100,
'PRIMER_INTERNAL_MAX_POLY_X': 100,
'PRIMER_SALT_MONOVALENT': 50.0,
'PRIMER_DNA_CONC': 50.0,
'PRIMER_MAX_NS_ACCEPTED': 0,
'PRIMER_MAX_SELF_ANY': 12,
'PRIMER_MAX_SELF_END': 8,
'PRIMER_PAIR_MAX_COMPL_ANY': 12,
'PRIMER_PAIR_MAX_COMPL_END': 8,
'PRIMER_PRODUCT_SIZE_RANGE': [
[75,100],[100,125],[125,150],
[150,175],[175,200],[200,225]
],
})
type(r)
dict
import json
result = json.dumps(r, indent=2)
print(result)
Output collapsed:
{
"PRIMER_LEFT_EXPLAIN": "considered 1997, too many Ns 25, GC content failed 32, low tm 1272, high tm 124, ok 544",
"PRIMER_RIGHT_EXPLAIN": "considered 1997, too many Ns 25, GC content failed 80, low tm 1366, high tm 108, high hairpin stability 10, ok 408",
"PRIMER_INTERNAL_EXPLAIN": "considered 3007, too many Ns 27, GC content failed 92, low tm 2608, high tm 11, high hairpin stability 18, ok 251",
"PRIMER_PAIR_EXPLAIN": "considered 823, unacceptable product size 808, no internal oligo 9, ok 6",
"PRIMER_LEFT_NUM_RETURNED": 5,
"PRIMER_RIGHT_NUM_RETURNED": 5,
"PRIMER_INTERNAL_NUM_RETURNED": 5,
"PRIMER_PAIR_NUM_RETURNED": 5,
"PRIMER_PAIR": [
{
"PENALTY": 1.373239688566116,
"COMPL_ANY_TH": 0.0,
"COMPL_END_TH": 0.0,
"PRODUCT_SIZE": 87,
"PRODUCT_TM": 81.3560105336193
},
{
"PENALTY": 1.5090296435631672,
"COMPL_ANY_TH": 0.0,
"COMPL_END_TH": 2.43414726309436,
"PRODUCT_SIZE": 94,
"PRODUCT_TM": 82.57648008607713
},
{
"PENALTY": 1.8643178301738885,
"COMPL_ANY_TH": 0.0,
"COMPL_END_TH": 0.0,
"PRODUCT_SIZE": 98,
"PRODUCT_TM": 82.41864248295076
},
{
"PENALTY": 1.9504613679555973,
"COMPL_ANY_TH": 0.0,
"COMPL_END_TH": 0.0,
"PRODUCT_SIZE": 85,
"PRODUCT_TM": 81.1771058683049
},
{
"PENALTY": 2.019318015989654,
"COMPL_ANY_TH": 0.0,
"COMPL_END_TH": 0.0,
"PRODUCT_SIZE": 88,
"PRODUCT_TM": 81.67536790039047
}
],
"PRIMER_LEFT": [
{
"PENALTY": 1.3299057711502655,
"SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"COORDS": [
46,
21
],
"TM": 59.670094228849734,
"GC_PERCENT": 52.38095238095238,
"SELF_ANY_TH": 10.513588697583486,
"SELF_END_TH": 10.513588697583486,
"HAIRPIN_TH": 42.52778282883122,
"END_STABILITY": 4.06
},
{
"PENALTY": 1.3299057711502655,
"SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"COORDS": [
46,
21
],
"TM": 59.670094228849734,
"GC_PERCENT": 52.38095238095238,
"SELF_ANY_TH": 10.513588697583486,
"SELF_END_TH": 10.513588697583486,
"HAIRPIN_TH": 42.52778282883122,
"END_STABILITY": 4.06
},
{
"PENALTY": 1.3299057711502655,
"SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"COORDS": [
46,
21
],
"TM": 59.670094228849734,
"GC_PERCENT": 52.38095238095238,
"SELF_ANY_TH": 10.513588697583486,
"SELF_END_TH": 10.513588697583486,
"HAIRPIN_TH": 42.52778282883122,
"END_STABILITY": 4.06
},
{
"PENALTY": 1.3299057711502655,
"SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"COORDS": [
46,
21
],
"TM": 59.670094228849734,
"GC_PERCENT": 52.38095238095238,
"SELF_ANY_TH": 10.513588697583486,
"SELF_END_TH": 10.513588697583486,
"HAIRPIN_TH": 42.52778282883122,
"END_STABILITY": 4.06
},
{
"PENALTY": 1.3299057711502655,
"SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"COORDS": [
46,
21
],
"TM": 59.670094228849734,
"GC_PERCENT": 52.38095238095238,
"SELF_ANY_TH": 10.513588697583486,
"SELF_END_TH": 10.513588697583486,
"HAIRPIN_TH": 42.52778282883122,
"END_STABILITY": 4.06
}
],
"PRIMER_RIGHT": [
{
"PENALTY": 0.043333917415850465,
"SEQUENCE": "TCTCCTCCTTAGCCTGCCTT",
"COORDS": [
132,
20
],
"TM": 59.95666608258415,
"GC_PERCENT": 55.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 0.0,
"END_STABILITY": 4.35
},
{
"PENALTY": 0.17912387241290162,
"SEQUENCE": "CAGTGCGTCTCCTCCTTAGC",
"COORDS": [
139,
20
],
"TM": 60.1791238724129,
"GC_PERCENT": 60.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 0.0,
"END_STABILITY": 3.09
},
{
"PENALTY": 0.534412059023623,
"SEQUENCE": "CATTCAGTGCGTCTCCTCCT",
"COORDS": [
143,
20
],
"TM": 59.46558794097638,
"GC_PERCENT": 55.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 0.0,
"END_STABILITY": 3.69
},
{
"PENALTY": 0.6205555968053318,
"SEQUENCE": "TCCTCCTTAGCCTGCCTTTG",
"COORDS": [
130,
20
],
"TM": 59.37944440319467,
"GC_PERCENT": 55.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 0.0,
"END_STABILITY": 2.77
},
{
"PENALTY": 0.6894122448393887,
"SEQUENCE": "GTCTCCTCCTTAGCCTGCCT",
"COORDS": [
133,
20
],
"TM": 60.68941224483939,
"GC_PERCENT": 60.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 0.0,
"END_STABILITY": 4.75
}
],
"PRIMER_INTERNAL": [
{
"PENALTY": 6.224608874676505,
"SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"COORDS": [
69,
24
],
"TM": 57.775391125323495,
"GC_PERCENT": 50.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 34.31335532251251
},
{
"PENALTY": 6.224608874676505,
"SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"COORDS": [
69,
24
],
"TM": 57.775391125323495,
"GC_PERCENT": 50.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 34.31335532251251
},
{
"PENALTY": 6.224608874676505,
"SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"COORDS": [
69,
24
],
"TM": 57.775391125323495,
"GC_PERCENT": 50.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 34.31335532251251
},
{
"PENALTY": 6.224608874676505,
"SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"COORDS": [
69,
24
],
"TM": 57.775391125323495,
"GC_PERCENT": 50.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 34.31335532251251
},
{
"PENALTY": 6.224608874676505,
"SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"COORDS": [
69,
24
],
"TM": 57.775391125323495,
"GC_PERCENT": 50.0,
"SELF_ANY_TH": 0.0,
"SELF_END_TH": 0.0,
"HAIRPIN_TH": 34.31335532251251
}
],
"PRIMER_PAIR_0_PENALTY": 1.373239688566116,
"PRIMER_LEFT_0_PENALTY": 1.3299057711502655,
"PRIMER_RIGHT_0_PENALTY": 0.043333917415850465,
"PRIMER_INTERNAL_0_PENALTY": 6.224608874676505,
"PRIMER_LEFT_0_SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"PRIMER_RIGHT_0_SEQUENCE": "TCTCCTCCTTAGCCTGCCTT",
"PRIMER_INTERNAL_0_SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"PRIMER_LEFT_0": [
46,
21
],
"PRIMER_RIGHT_0": [
132,
20
],
"PRIMER_INTERNAL_0": [
69,
24
],
"PRIMER_LEFT_0_TM": 59.670094228849734,
"PRIMER_RIGHT_0_TM": 59.95666608258415,
"PRIMER_INTERNAL_0_TM": 57.775391125323495,
"PRIMER_LEFT_0_GC_PERCENT": 52.38095238095238,
"PRIMER_RIGHT_0_GC_PERCENT": 55.0,
"PRIMER_INTERNAL_0_GC_PERCENT": 50.0,
"PRIMER_LEFT_0_SELF_ANY_TH": 10.513588697583486,
"PRIMER_RIGHT_0_SELF_ANY_TH": 0.0,
"PRIMER_INTERNAL_0_SELF_ANY_TH": 0.0,
"PRIMER_LEFT_0_SELF_END_TH": 10.513588697583486,
"PRIMER_RIGHT_0_SELF_END_TH": 0.0,
"PRIMER_INTERNAL_0_SELF_END_TH": 0.0,
"PRIMER_LEFT_0_HAIRPIN_TH": 42.52778282883122,
"PRIMER_RIGHT_0_HAIRPIN_TH": 0.0,
"PRIMER_INTERNAL_0_HAIRPIN_TH": 34.31335532251251,
"PRIMER_LEFT_0_END_STABILITY": 4.06,
"PRIMER_RIGHT_0_END_STABILITY": 4.35,
"PRIMER_PAIR_0_COMPL_ANY_TH": 0.0,
"PRIMER_PAIR_0_COMPL_END_TH": 0.0,
"PRIMER_PAIR_0_PRODUCT_SIZE": 87,
"PRIMER_PAIR_0_PRODUCT_TM": 81.3560105336193,
"PRIMER_PAIR_1_PENALTY": 1.5090296435631672,
"PRIMER_LEFT_1_PENALTY": 1.3299057711502655,
"PRIMER_RIGHT_1_PENALTY": 0.17912387241290162,
"PRIMER_INTERNAL_1_PENALTY": 6.224608874676505,
"PRIMER_LEFT_1_SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"PRIMER_RIGHT_1_SEQUENCE": "CAGTGCGTCTCCTCCTTAGC",
"PRIMER_INTERNAL_1_SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"PRIMER_LEFT_1": [
46,
21
],
"PRIMER_RIGHT_1": [
139,
20
],
"PRIMER_INTERNAL_1": [
69,
24
],
"PRIMER_LEFT_1_TM": 59.670094228849734,
"PRIMER_RIGHT_1_TM": 60.1791238724129,
"PRIMER_INTERNAL_1_TM": 57.775391125323495,
"PRIMER_LEFT_1_GC_PERCENT": 52.38095238095238,
"PRIMER_RIGHT_1_GC_PERCENT": 60.0,
"PRIMER_INTERNAL_1_GC_PERCENT": 50.0,
"PRIMER_LEFT_1_SELF_ANY_TH": 10.513588697583486,
"PRIMER_RIGHT_1_SELF_ANY_TH": 0.0,
"PRIMER_INTERNAL_1_SELF_ANY_TH": 0.0,
"PRIMER_LEFT_1_SELF_END_TH": 10.513588697583486,
"PRIMER_RIGHT_1_SELF_END_TH": 0.0,
"PRIMER_INTERNAL_1_SELF_END_TH": 0.0,
"PRIMER_LEFT_1_HAIRPIN_TH": 42.52778282883122,
"PRIMER_RIGHT_1_HAIRPIN_TH": 0.0,
"PRIMER_INTERNAL_1_HAIRPIN_TH": 34.31335532251251,
"PRIMER_LEFT_1_END_STABILITY": 4.06,
"PRIMER_RIGHT_1_END_STABILITY": 3.09,
"PRIMER_PAIR_1_COMPL_ANY_TH": 0.0,
"PRIMER_PAIR_1_COMPL_END_TH": 2.43414726309436,
"PRIMER_PAIR_1_PRODUCT_SIZE": 94,
"PRIMER_PAIR_1_PRODUCT_TM": 82.57648008607713,
"PRIMER_PAIR_2_PENALTY": 1.8643178301738885,
"PRIMER_LEFT_2_PENALTY": 1.3299057711502655,
"PRIMER_RIGHT_2_PENALTY": 0.534412059023623,
"PRIMER_INTERNAL_2_PENALTY": 6.224608874676505,
"PRIMER_LEFT_2_SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"PRIMER_RIGHT_2_SEQUENCE": "CATTCAGTGCGTCTCCTCCT",
"PRIMER_INTERNAL_2_SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"PRIMER_LEFT_2": [
46,
21
],
"PRIMER_RIGHT_2": [
143,
20
],
"PRIMER_INTERNAL_2": [
69,
24
],
"PRIMER_LEFT_2_TM": 59.670094228849734,
"PRIMER_RIGHT_2_TM": 59.46558794097638,
"PRIMER_INTERNAL_2_TM": 57.775391125323495,
"PRIMER_LEFT_2_GC_PERCENT": 52.38095238095238,
"PRIMER_RIGHT_2_GC_PERCENT": 55.0,
"PRIMER_INTERNAL_2_GC_PERCENT": 50.0,
"PRIMER_LEFT_2_SELF_ANY_TH": 10.513588697583486,
"PRIMER_RIGHT_2_SELF_ANY_TH": 0.0,
"PRIMER_INTERNAL_2_SELF_ANY_TH": 0.0,
"PRIMER_LEFT_2_SELF_END_TH": 10.513588697583486,
"PRIMER_RIGHT_2_SELF_END_TH": 0.0,
"PRIMER_INTERNAL_2_SELF_END_TH": 0.0,
"PRIMER_LEFT_2_HAIRPIN_TH": 42.52778282883122,
"PRIMER_RIGHT_2_HAIRPIN_TH": 0.0,
"PRIMER_INTERNAL_2_HAIRPIN_TH": 34.31335532251251,
"PRIMER_LEFT_2_END_STABILITY": 4.06,
"PRIMER_RIGHT_2_END_STABILITY": 3.69,
"PRIMER_PAIR_2_COMPL_ANY_TH": 0.0,
"PRIMER_PAIR_2_COMPL_END_TH": 0.0,
"PRIMER_PAIR_2_PRODUCT_SIZE": 98,
"PRIMER_PAIR_2_PRODUCT_TM": 82.41864248295076,
"PRIMER_PAIR_3_PENALTY": 1.9504613679555973,
"PRIMER_LEFT_3_PENALTY": 1.3299057711502655,
"PRIMER_RIGHT_3_PENALTY": 0.6205555968053318,
"PRIMER_INTERNAL_3_PENALTY": 6.224608874676505,
"PRIMER_LEFT_3_SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"PRIMER_RIGHT_3_SEQUENCE": "TCCTCCTTAGCCTGCCTTTG",
"PRIMER_INTERNAL_3_SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"PRIMER_LEFT_3": [
46,
21
],
"PRIMER_RIGHT_3": [
130,
20
],
"PRIMER_INTERNAL_3": [
69,
24
],
"PRIMER_LEFT_3_TM": 59.670094228849734,
"PRIMER_RIGHT_3_TM": 59.37944440319467,
"PRIMER_INTERNAL_3_TM": 57.775391125323495,
"PRIMER_LEFT_3_GC_PERCENT": 52.38095238095238,
"PRIMER_RIGHT_3_GC_PERCENT": 55.0,
"PRIMER_INTERNAL_3_GC_PERCENT": 50.0,
"PRIMER_LEFT_3_SELF_ANY_TH": 10.513588697583486,
"PRIMER_RIGHT_3_SELF_ANY_TH": 0.0,
"PRIMER_INTERNAL_3_SELF_ANY_TH": 0.0,
"PRIMER_LEFT_3_SELF_END_TH": 10.513588697583486,
"PRIMER_RIGHT_3_SELF_END_TH": 0.0,
"PRIMER_INTERNAL_3_SELF_END_TH": 0.0,
"PRIMER_LEFT_3_HAIRPIN_TH": 42.52778282883122,
"PRIMER_RIGHT_3_HAIRPIN_TH": 0.0,
"PRIMER_INTERNAL_3_HAIRPIN_TH": 34.31335532251251,
"PRIMER_LEFT_3_END_STABILITY": 4.06,
"PRIMER_RIGHT_3_END_STABILITY": 2.77,
"PRIMER_PAIR_3_COMPL_ANY_TH": 0.0,
"PRIMER_PAIR_3_COMPL_END_TH": 0.0,
"PRIMER_PAIR_3_PRODUCT_SIZE": 85,
"PRIMER_PAIR_3_PRODUCT_TM": 81.1771058683049,
"PRIMER_PAIR_4_PENALTY": 2.019318015989654,
"PRIMER_LEFT_4_PENALTY": 1.3299057711502655,
"PRIMER_RIGHT_4_PENALTY": 0.6894122448393887,
"PRIMER_INTERNAL_4_PENALTY": 6.224608874676505,
"PRIMER_LEFT_4_SEQUENCE": "GCATCAGTGAGTACAGCATGC",
"PRIMER_RIGHT_4_SEQUENCE": "GTCTCCTCCTTAGCCTGCCT",
"PRIMER_INTERNAL_4_SEQUENCE": "ACTGGAAGAGAGGGTCATGCAACA",
"PRIMER_LEFT_4": [
46,
21
],
"PRIMER_RIGHT_4": [
133,
20
],
"PRIMER_INTERNAL_4": [
69,
24
],
"PRIMER_LEFT_4_TM": 59.670094228849734,
"PRIMER_RIGHT_4_TM": 60.68941224483939,
"PRIMER_INTERNAL_4_TM": 57.775391125323495,
"PRIMER_LEFT_4_GC_PERCENT": 52.38095238095238,
"PRIMER_RIGHT_4_GC_PERCENT": 60.0,
"PRIMER_INTERNAL_4_GC_PERCENT": 50.0,
"PRIMER_LEFT_4_SELF_ANY_TH": 10.513588697583486,
"PRIMER_RIGHT_4_SELF_ANY_TH": 0.0,
"PRIMER_INTERNAL_4_SELF_ANY_TH": 0.0,
"PRIMER_LEFT_4_SELF_END_TH": 10.513588697583486,
"PRIMER_RIGHT_4_SELF_END_TH": 0.0,
"PRIMER_INTERNAL_4_SELF_END_TH": 0.0,
"PRIMER_LEFT_4_HAIRPIN_TH": 42.52778282883122,
"PRIMER_RIGHT_4_HAIRPIN_TH": 0.0,
"PRIMER_INTERNAL_4_HAIRPIN_TH": 34.31335532251251,
"PRIMER_LEFT_4_END_STABILITY": 4.06,
"PRIMER_RIGHT_4_END_STABILITY": 4.75,
"PRIMER_PAIR_4_COMPL_ANY_TH": 0.0,
"PRIMER_PAIR_4_COMPL_END_TH": 0.0,
"PRIMER_PAIR_4_PRODUCT_SIZE": 88,
"PRIMER_PAIR_4_PRODUCT_TM": 81.67536790039047
}
seq = ('GCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCCTACATTTT'
'AGCATCAGTGAGTACAGCATGCTTACTGGAAGAGAGGGTCATGCA'
'ACAGATTAGGAGGTAAGTTTGCAAAGGCAGGCTAAGGAGGAGACG'
'CACTGAATGCCATGGTAAGAACTCTGGACATAAAAATATTGGAAG'
'TTGTTGAGCAAGTNAAAAAAATGTTTGGAAGTGTTACTTTAGCAA'
'TGGCAAGAATGATAGTATGGAATAGATTGGCAGAATGAAGGCAAA'
'ATGATTAGACATATTGCATTAAGGTAAAAAATGATAACTGAAGAA'
'TTATGTGCCACACTTATTAATAAGAAAGAATATGTGAACCTTGCA'
'GATGTTTCCCTCTAGTAG')
seq
'GCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCCTACATTTTAGCATCAGTGAGTACAGCATGCTTACTGGAAGAGAGGGTCATGCAACAGATTAGGAGGTAAGTTTGCAAAGGCAGGCTAAGGAGGAGACGCACTGAATGCCATGGTAAGAACTCTGGACATAAAAATATTGGAAGTTGTTGAGCAAGTNAAAAAAATGTTTGGAAGTGTTACTTTAGCAATGGCAAGAATGATAGTATGGAATAGATTGGCAGAATGAAGGCAAAATGATTAGACATATTGCATTAAGGTAAAAAATGATAACTGAAGAATTATGTGCCACACTTATTAATAAGAAAGAATATGTGAACCTTGCAGATGTTTCCCTCTAGTAG'
len(seq)
378